单细胞文章解析(3):单核RNA和ATAC测序揭示在单细胞水平上染色质可及性对拟南芥根基因表达的影响
Single-nucleus RNA and ATAC sequencing reveals the impact of chromatin accessibility on gene expression inArabidopsisroots at the singlecell level
一、研究概述
和其他复杂的生物体一样,植物同样是由不同的细胞类型组成的。不同的细胞类型具有其独特的生物学功能,生物学功能的差异使细胞类群建立起特异的转录特性。为了深入了解植物基因表达调控机制,本文作者使用**single-nucleus RNA sequencing (sNucRNA-seq)和single-nucleus assay for transposase accessible chromatin sequencing (sNucATACseq)**两种单细胞技术应用于拟南芥根的研究中。通过将本文的单核转录组数据与已发表的原生质体转录组数据进行比较,证明了利用细胞核作为生物实体来建立植物细胞类型特异性转录组的有效性。此外,本文的sNucRNA-seq结果揭示了scRNA-seq无法捕获到的细胞类型的转录组。本文使用sNucATAC-seq数据同样将细胞核分配到不同的类群中,该结果说明不同细胞类群之间具有较为差异的染色质可及性。为了揭示染色质可及性对基因表达的影响,本文整合了sNucRNA-seq和sNucATAC-seq数据,证明了细胞类型特异性标记基因显示了细胞类型特异性染色质可及性模式。数据表明,不同的染色质可及性是在细胞类型水平上调节基因活性的关键机制。
二、研究背景
组成植物的各种细胞类型获得独特的生物学功能,取决于它们对相同基因组信息的不同利用,以产生细胞类型特异性的转录图谱。细胞和细胞类型对基因组信息的不同利用被认为部分依赖于不同的染色质可及性。染色质纤维结构的变化影响调控打包对基因组DNA的可及性,如转录因子(TFs)。这一说法得到了人类ENCODE项目的支持,该项目最近发现,在单细胞水平上染色质图谱的建立对于揭示预测的tf结合位点具有高度的信息量。最终,每个植物细胞都会激活或抑制特定的基因,以实现其细胞类型和对环境信号的响应所固有的生物功能。在动物科学领域,单细胞RNA测序(scRNA-seq)和单细胞ATAC-seq技术已成功应用于各种细胞类型和组织,以更好地理解染色质动态可及性对基因表达的影响。
最近,scRNA-seq方法已被应用于拟南芥根原生质体,允许准确表征数千个细胞的转录谱及其在突变体或应激反应中的差异调控。这些研究揭示了单细胞技术在建立各种拟南芥根细胞和细胞类型的转录组图和细胞发育过程中基因表达的动态调控的力量。然而,利用植物原生质体作为生物实体来分析基因表达存在一些问题。例如,一些细胞类型的细胞壁不容易被消化处理,原生质体过程本身对基因表达有重要影响,还有一种倾向于对较小尺寸的细胞/原生质体进行测序。此外,有效分离植物原生质体需要开发特定的细胞壁降解酶,为存在于不同植物种类的细胞壁的不同生化成分、细胞的发育阶段(即,初级细胞壁和次级细胞壁的生化组成差异)和根细胞的相对位置(即外部和内部位置)。
作为原生质体的替代品,大量的植物细胞的核被用来从植物细胞中获取转录组信息。例如,通过细胞核标记分离技术建立起水稻根、拟南芥胚胎和种子胚乳的转录组图谱。然而,这些方法也有各种局限性。例如,使用细胞核标记分离技术的前提是鉴定出表达报告基因的细胞类型特异性标记基因,并需要产生转基因物质。
为了克服一些与植物原生质体的制备和操作过程中存在的一些问题,作者使用分离的植物核来建立数千个植物单细胞的转录组。以往在各种动物系统上进行的单核转录组实验表明,与分离细胞相比,细胞核同样可以用来建立具有生物学意义的转录组信息。我们的数据揭示了细胞核和原生质体转录组之间的高度相似性,并发现了额外的根细胞型转录组,支持使用分离细胞核作为有价值的生物实体来获取单细胞基因表达。为了更深入地了解拟南芥根细胞和细胞类型中基因表达的调控机制,作者开发并应用了单核ATAC-seq (sNucATAC-seq)。sNucATAC-seq揭示了细胞类型特异的染色质可及性以及在细胞分化和发育过程中染色质可及性发生的动态变化。在建立了拟南芥根主要细胞类型的独特和保守的染色质可及性图谱后,作者整合了sNucRNA-seq和sNucATAC-seq数据集,以突出染色质可及性对拟南芥基因活性的影响。
三、结果解读
3.1 拟南芥单核转录组图谱的构建
为了准确比较sNucRNA-seq和scRNA-seq转录组,作者种植了拟南芥幼苗,并按照Ryu等人(2019)的描述分离了主根。本文进行了五次生物学重复,最终得到了10,548个细胞核。每个细胞核中表达的基因数量的中位数是1124,所有细胞核总共检测到24,510个基因,占拟南芥基因组总基因数目的89.4%。相比之下,Ryu等人(2019)选取的7437个拟南芥原生质体的转录组数据中每个细胞检测到4739个表达基因和25 177个总基因数(91.8%)。由于细胞质相对于细胞核中会积累更多的转录本,而且更为稳定性,所以相对于细胞核,每个原生质体中会鉴定出较多的表达基因数量。这一结果还表明,核转录组代表了基因动态转录活性的快照,而细胞转录组可能代表了基因活性随时间的整合。
为了评估从sNucRNA-seq数据中获得的核转录组的生物学意义,作者对完整根、原生质体悬浮液的bulk RNA数据,以及scRNA-seq和sNucRNA-seq(本文章)的pseudo-bulk RNA-seq进行了相关性分析。其中完整根的转录组与原生质体的转录组具有较高的相关性,与pseudo-bulk scRNA-seq转录组数据相关性在0.859到0.892之间,与pseudo-bulk sNucRNA-seq转录组数据的相关性为0.879。在排除了346个已知原生质体响应基因后,他们之间的相关性都有所增加。这些结果表明,拟南芥根的sNucRNA-seq转录组和原生质体转录组一样与整个根的转录组具有高度的相关性。总体而言,通过拟南芥根的细胞核生成的基于单核的转录组很好地反映了传统方法生成的根转录组。
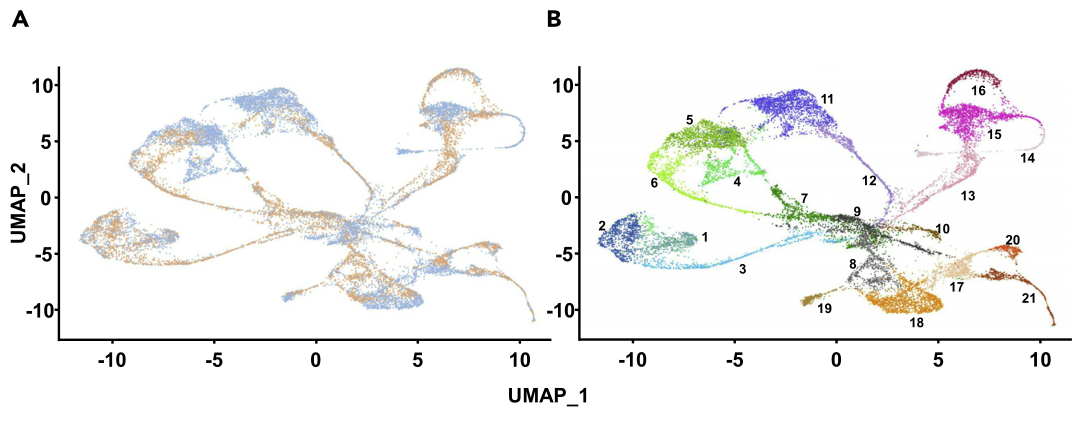
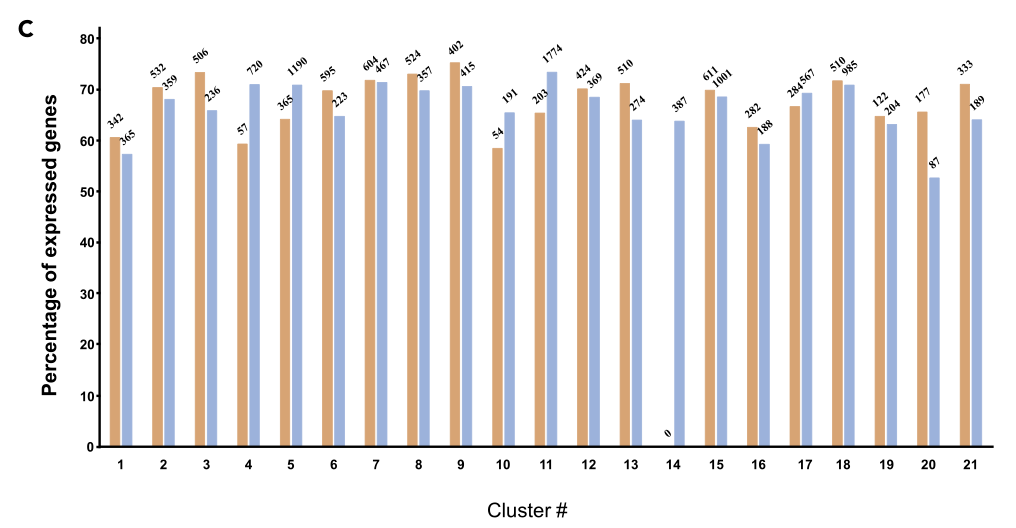
通过Seurat软件,作者将本文章中的10,548个拟南芥根细胞核与7437个拟南芥根原生质体进行整合,分别得到21和20个细胞类群。UMAP显示,其中20个细胞类群是是有重叠的,在4和11类群中89%都是有细胞核组成的,14类群全部都是由细胞核组成的。在拟南芥基因组中有73.7%的基因被sNucRNA-seq和scRNA-seq技术共同检测到的。除了Cluster 14,sNucRNA-seq类群中表达基因所占的比例(52.7% to 73.3%)与scRNA-seq类群中的表达基因所占的比例(58.4% to 75.2%)之间的差异是不显著的。这些结果表明,细胞和核转录组提供了相似的转录组信息,表明分离的植物核可以在单细胞类型水平上建立有意义的转录组信息。通过识别三个新的细胞类群,本文的数据还表明,sNucRNA-seq方法捕获了比scRNA-seq更多样化和更有代表性的拟南芥根细胞类型群体。
3.2 细胞类群的注释
利用最近发表的拟南芥根单细胞转录组和根基因表达与调控的相关信息,本文共收集了101个细胞类型的marker基因。通过观察这些marker基因在这21个细胞类群中的表达富集程度,最终这21个细胞类群被分为了6个主要的类群:trichoblasts (clusters 1–3), atrichoblasts (clusters 4–7), meristematic cells (clusters 8–10), cortical cells (clusters 11 and 12), endodermal cells (clusters 13–16) and stele cells (clusters 17–21)。此外,依据marker基因,19和21号类群被分别定义为韧皮部和木质部。
接下来文章重点对sNucRNA-seq细胞群(clusters 14)进行了详细的分析。Cluster 14依据内皮层marker基因和皮层marker基因被分成了两个亚群,cluster 14a和cluster 14b。
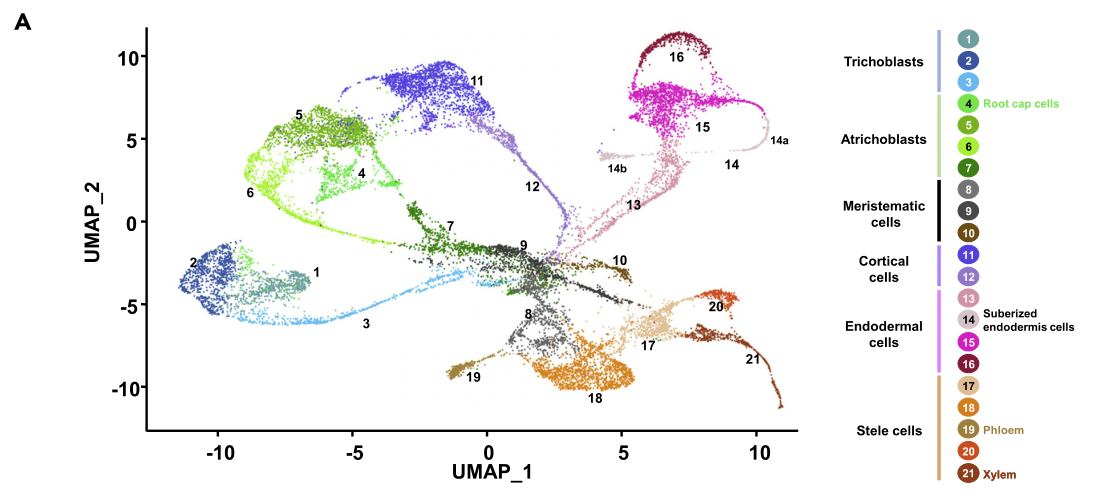
3.2.1 Cluster 14a
Cluster 14a中特异性表达过氧化物酶和GDSL-motif基因。GDSL家族的成员,如水稻WDL1和番茄GDSL1基因,控制着细胞分化的过程。因此cluster 14a可能是由分化了的细胞组成。这一假设得到了根过氧化物酶在控制活性氧的产生来调节细胞伸长和分化中的作用的支持。更有证据显示,,UPBEAT1基因(AT2G47270),过氧化物酶基因的转录活性的主要抑制因子和活性氧物种分布,并且该基因负调控分生组织大小,除聚类4、10、11和14外,其余均广泛表达。除了控制细胞分化,GDSL脂肪酶在角质生物合成中也发挥着重要作用。通过对sc/sNucRNAseq数据集的挖掘,我们发现了许多其他基因,它们优先表达于14a聚类,并参与软木脂和角质的生物合成。此前的研究表明,木脂素和角质在侧根出现的位置和内皮层分化期间强烈沉积。综上所述,UPBEAT1、编码过氧化物酶的几个基因、GDSL基因和其他与皮层和内胚层标记基因活性相关的木脂质/角质生物合成相关基因的转录模式,提示群集14a的细胞是分化的内皮层细胞,其特征是细胞壁细分。14a群细胞的转录组特征来自于分离的细胞核而不是分离的原生质体,这可能是由于它们的细胞壁消化率低,这是木脂质和角质积累的结果。
3.2.2 Cluster 14b
在Cluster 14b亚群中,皮层细胞特异表达的基因在该亚群中富集。SCRAMBLED/STRUBBELIG (SCM, AT1G11130)基因在根表皮细胞的形成过程中起关键作用。在Cluster 14b中,参与脂质代谢的几个基因的表达进一步支持了表达SCM细胞的特异性聚类。膜脂重塑也被证明在根毛细胞分化中发挥关键作用。综上所述,推测构成簇14b的皮层细胞在拟南芥表皮根细胞的分化和模式形成中发挥作用。
3.2.3 其他信息
Cluster 14b中CEP1 (AT5G50260)和EXI1 (AT2G14095)富集表达,这两个基因以前被认为是根冠细胞死亡程序的调控因子。此外,还鉴定了在花发育过程中控制细胞死亡的基因KIRA1 (AT4g28530)。其他的细胞死亡marker基因(如BFN1, RNS3, SCPL48, DMP4,和PASPA3)也大多在木质部类群4和21的一个子集中表达。以往的研究表明,细胞死亡程序在木质部和根冠的发育中起着至关重要的作用。以往的研究表明,细胞死亡程序在木质部和根冠的发育中起关键作用,支持这些细胞死亡标记基因在木质部21簇中的特异性活性,并将组成4簇的细胞定位为根冠细胞。
3.3 单细胞分辨率ATAC-seq揭示了染色质可及性对基因表达的影响
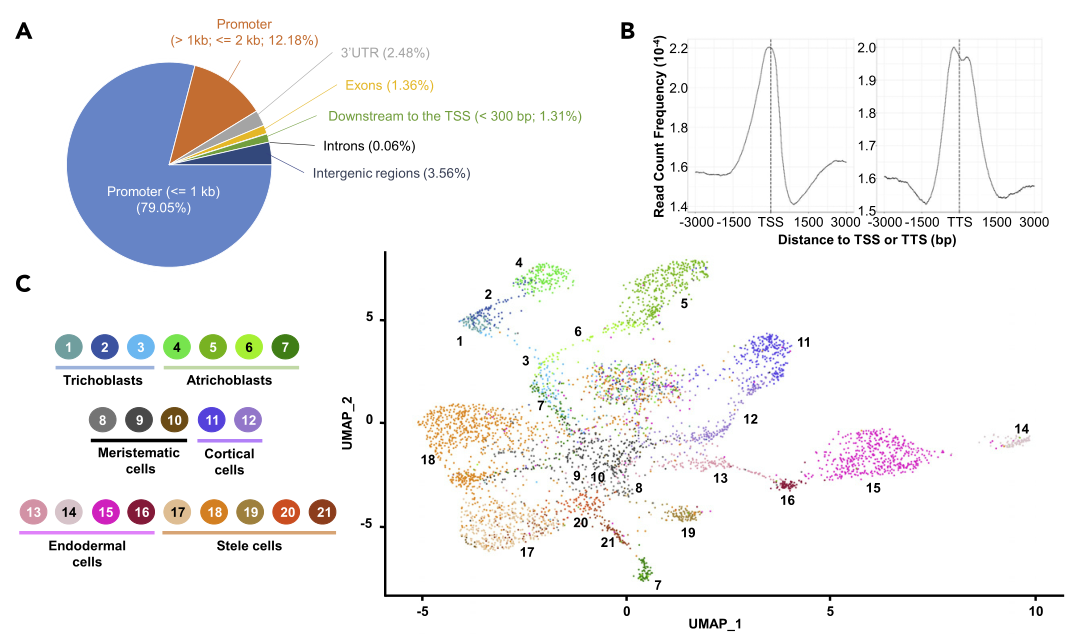
虽然体细胞之间的基因组信息几乎是相同的(即体细胞突变除外),但为了通过细胞类型特异性转录基因调控实现其独特的生物学功能,需要对其进行不同的利用,特别是通过细胞间染色质可及性的差异。迄今为止,bulk RNA-和ATAC-seq数据集显示出低相关性(Maher et al., 2018),这可能是所使用样本的细胞异质性的结果。这一假设得到了人类ENCODE项目的支持,该项目最近发现,在单细胞水平上染色质图谱的建立对于揭示预测的转录因子结合位点具有高度的信息量。为了更好地评价染色质可及性在控制植物细胞和细胞类型之间的基因表达方面的影响,将10X Genomics sNucATAC-seq技术应用于从两个独立的生物复制中分离的拟南芥根核。在这6768个核中,有4764个通过了下游的质量控制过滤器。
通过序列比对,本文每个细胞核大约有10,253个DNA fragments,共有20,803个染色质可及性位点鉴定出来。作为比较,Lu et al.(2017)、Tannenbaum et al.(2018)和Maher et al.(2018)分别从拟南芥幼苗和根的bulk ATAC-seq分析中确定了约2万个和4万个染色质可及性位点。在这20,803个染色质可及性位点中,分别鉴定出3487个和15,730个具有细胞型特异性Peaks和”static” Peaks。作者观察到染色质可及性位点大多位于含有顺式调控元件的转录起始位点(TSSs)上游1000 bp的窗口内和基因的转录终止位点(TTSs)附近。为了证明本文sNucATAC-seq数据的可靠性,作者发现来自完整的拟南芥根尖的bulk ATAC-seq (Maher et al., 2018)和本文的pseudo-bulked sNucATAC-seq数据集(即,SRCC = 0.95)之间存在高度相关性。
考虑到染色质位点的可达性是促进基因表达的前提条件,作者认为位于TSS附近的细胞型特异性ATAC-seq Peaks有助于调控细胞型标记基因的表达。因此,利用Signac软件整合sNucATAC-seq和sc/sNucRNA-seq分析,在细胞类型特异性环境中表达的基因的TSS附近寻找开放染色质。最终,构建了21个sNucATAC-seq细胞类群。为了评估使用sc/sNucRNA-seq数据集根据拟南芥核的染色质可及性特征聚类的影响,我们根据核的开放染色质差异峰对核进行了de novo聚类。这种方法再次导致了21个聚类的鉴定,这些聚类与我们整合的sNucATAC-seq和sc/ sNucRNA-seq分析鉴定的聚类稍有差异。这一结果表明,染色质的可及性足以揭示拟南芥根细胞的复杂性。
首先,sNucATAC-seq Clusters 1、2和3;Clusters 4、5和6;Clusters 11和12;Clusters 13、14、15和16;Clusters17、18、19、20和21,分别由trichoblasts, the atrichoblasts, and the cortical, endodermal, and stele cells组成,该聚类结果类似于sNucRNA-seq的分析结果。其次,类似与sc/sNucRNA-seq的UMAP拓扑结构,sNuc-ATAC-seq的聚类结果中,未分化细胞类群(例如,Clusters 8、9和10)位于UMAP的中心,而已分化的细胞类群位于它的外围。总体上相似的地形表明,染色质可及性和基因表达都可以作为分子标记来注释植物细胞类型,并支持某些基因的染色质可及性与其转录活性之间的相关性。
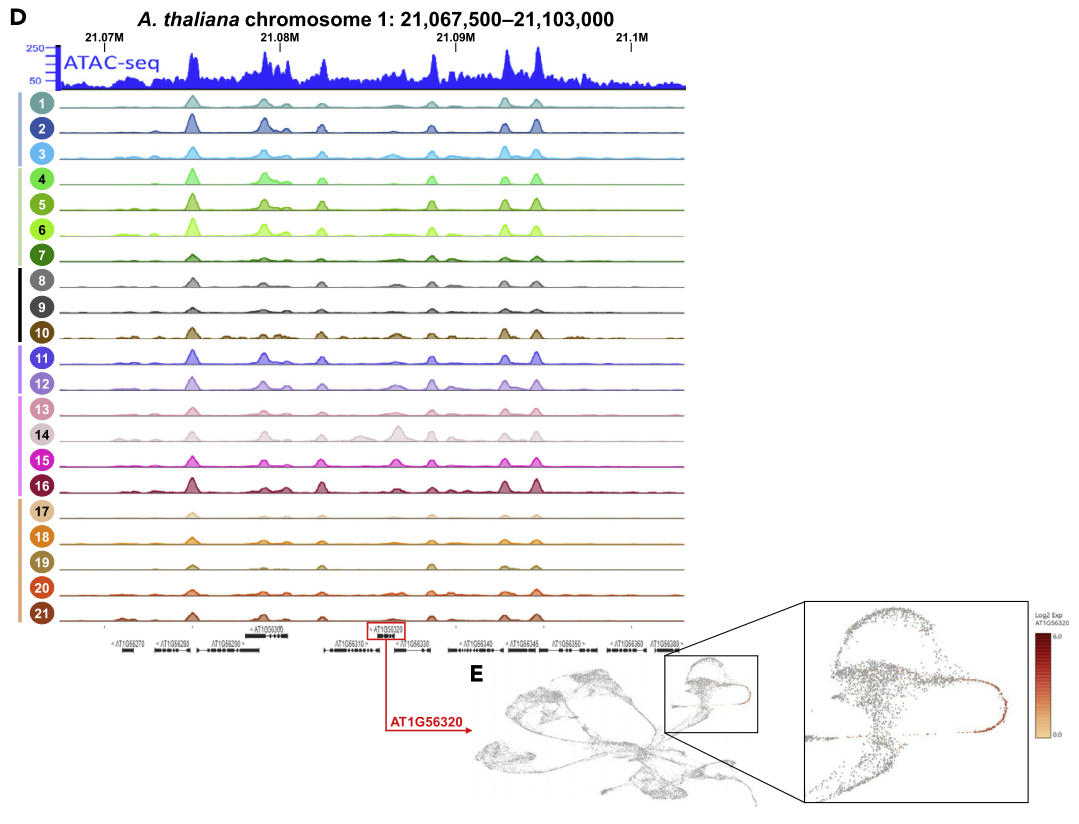
为了说明sNucATAC-seq测序技术相对于bulk ATAC-seq可以获得更高的分辨率,并且可以获得染色质可及性离散的变化,作者首先比较了chr1:21 067 500–21 103 000(Tannenbaum et al., 2018)这一位置的ATAC信号分布。可以看出,sNucATAC-seq的21个细胞类群中可以清楚的识别出bulk ATAC-seq技术揭示的相同的主要Peaks。此外,sNucATAC-seq鉴定出了只在部分细胞类群中出现的Peaks(e.g., ATAC-seq peak located in the promoter region of AT1G56320 in the sNucATAC-seq clusters 14 and 15)。上述分析结果表明,单细胞分辨率的ATAC-seq分析有潜力揭示可接近染色质的离散和细胞类型特异性位点。
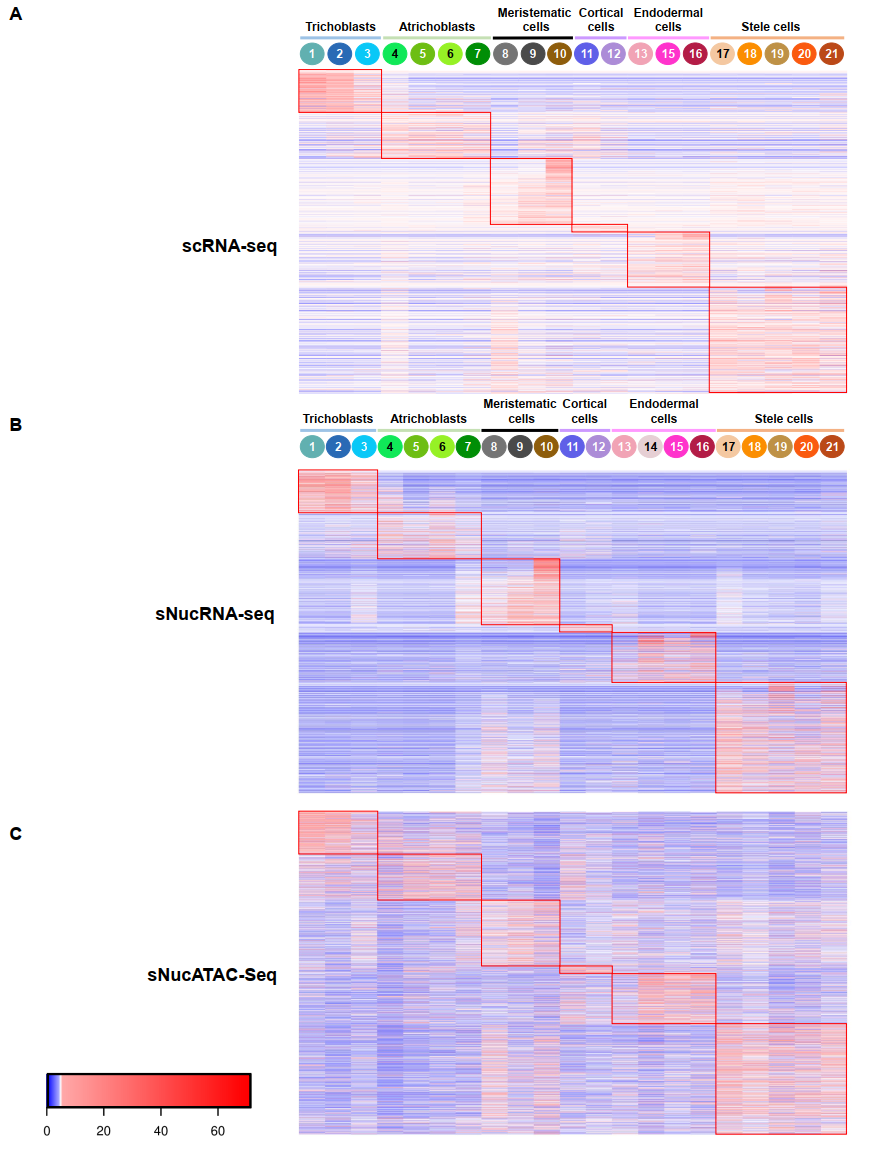
作者进行了进一步的分析,以揭示细胞型染色质可及性如何在控制基因表达中发挥关键作用。例如,热图显示2756个在拟南芥毛细胞、成纤维细胞和分生组织、皮层、内胚层和中柱细胞中优先表达的基因,他们的聚类模式相似,这一结果表明染色质可及性与基因活性之间存在一定的相关性,至少对这些细胞类型偏好基因来说是这样的。
因此,为了进一步揭示sc/sNucRNA-seq和sNucATAC-seq实验之间的对应关系,作者进行了拟南芥标记基因表达与TSS位点染色质可及性之间的相关性分析。通过对scRNA-seq和sNucRNA-seq数据集的挖掘,根据每个聚类中与其他聚类相比的表达倍数变化和它们的最低P值,从每个聚类中选择前20个标记基因。由于集群之间存在一些冗余,鉴定了370个独特的标记基因,其中32个属于用于注释sc/sNucRNA-seq类群的101个标记基因。这些基因中,有336个基因的TSS附近有至少一个sNucATAC-seq Peak。作为比较,作者同样将相同的分析方法用于811个housekeeping (HK)基因和三组随机挑选的336个基因中。结果显示,marker基因具有更好的相关性。这一结果表明,组成相同组织的细胞在某种程度上具有相似的转录组和表观基因组特征,这可能是实现其组织特异性生物学功能所必需的。基因表达和染色质可及性之间的显著相关性表明,核小体在基因组DNA双链上靠近基因TSS的位置,在控制至少一部分标记基因的活性方面起着关键作用。
3.4 ATAC-seq在单细胞分辨率上的染色质可及性可以作为根毛和内胚层细胞发育状态的分子标记
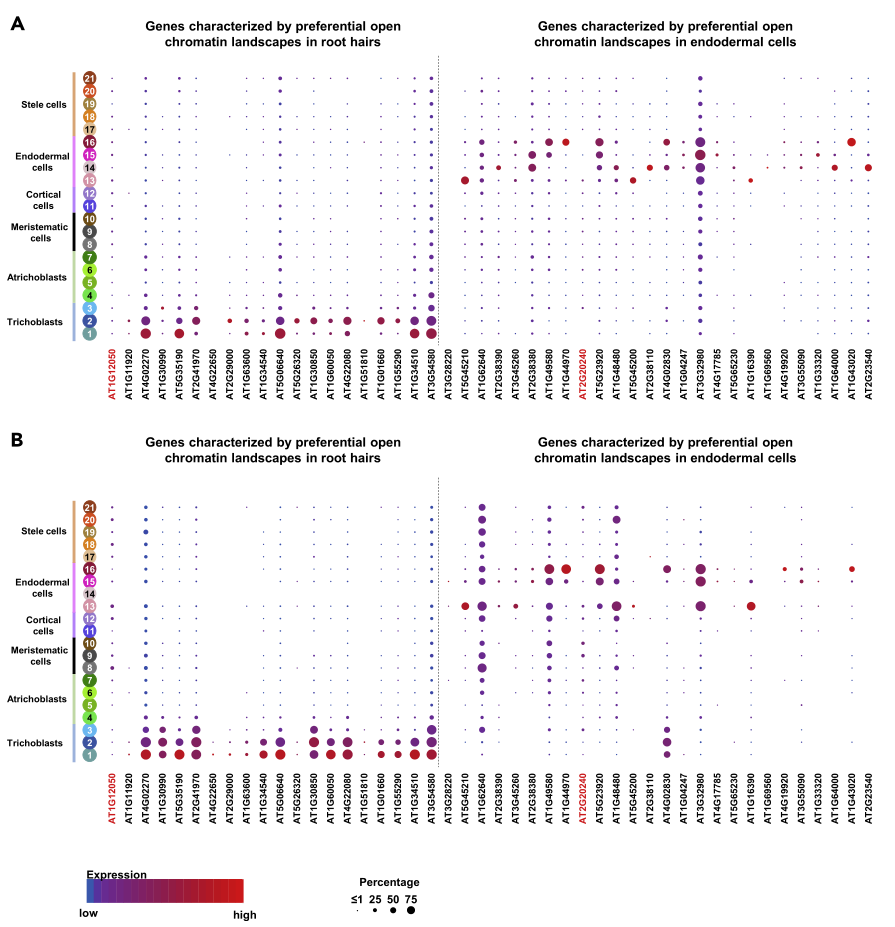
基于标记基因的表达谱注释植物单细胞类型(Denyer et al., 2019;Jean-Baptiste et al., 2019;Ryu et al., 2019;Shulse et al., 2019;Zhang et al., 2019)。由于基因表达与染色质可及性之间的相关性,因此,后者也可以用来注释拟南芥根的细胞类型。这一假设得到了对各种动物器官和选定植物细胞进行的类似分析的支持(Preissl et al., 2018;Sijacic et al., 2018;Sinnamon等人,2019)。为了进一步验证这一假设,作者利用本文的单细胞分辨率ATAC-seq数据集,集中分析了成熟拟南芥根毛和内胚层细胞的细胞类群。在11 858个与拟南芥TSSs配对的sNucATAC-seq峰中,分别有20个和26个在根毛和内胚层簇中被特异性识别。通过对scRNA-seq和sNucRNAseq数据集的挖掘,我们分别鉴定出19个(95%)和25个(96.2%)基因在拟南芥根毛和内胚层细胞中优先表达。我们首先通过识别聚类1、2、3和聚类13、14、15、16的可接近染色质峰值来进行分析,从而识别了相关的根毛和内胚层标记基因。除了支持染色质可及性在控制植物细胞基因表达中的作用外,这项工作还强调染色质可及性可作为一种分子标记来注释特定的细胞类型。
植物核可以用来捕获各种细胞类型的转录组。这种方法将为探索细胞分化和伸长过程中以及细胞类型之间的动态转录组变化开辟新的途径。当应用于各种植物物种时,基于单核的转录组学和表观基因组学分析也将使植物物种之间更有针对性的比较分析,以揭示单细胞型水平上的基因表达的进化。
四、总结
- 首先,作者比较了scRNA-seq与sNucRNA-seq之间的相关性,证明了sNucRNA-seq的有效性。最终通过整合两类数据得到了21个细胞类群
- 通过收集的细胞marker基因对细胞类群进行了注释。重点分析了Cluster 14细胞类群。
- 通过整合sc/sNucRNA-seq与sNucATAC-seq数据,作者得到了与转录组相似的拓扑结构
- 通过特殊的例子,作者证明了sNucATAC-seq相较于bulk ATAC-seq具有更好的分辨率,可以更为精确的反应染色质可及性的离散性和特异性。
- 为了进一步揭示sc/sNucRNA-seq和sNucATAC-seq实验之间的对应关系,作者进行了拟南芥标记基因表达与TSS位点染色质可及性之间的相关性分析。
- 最后,通过实验数据说明了,sNucATAC-seq中的Peaks同样可以作为生物学marker来对细胞类型进行注释。
单细胞文章解析(3):单核RNA和ATAC测序揭示在单细胞水平上染色质可及性对拟南芥根基因表达的影响

